

INTRODUCTION
Colorectal cancer (CRC) is a major cause of cancer-related deaths in developed countries, and its incidence is also rapidly increasing in developing countries [1]. The mortality associated with CRC has decreased because of early detection using advanced colonoscopy screening and the use of anticancer drugs such as irinotecan as first-line therapy, and molecular targeted therapy [2]. However, strategies for CRC management are limited, especially in the advanced stages and the increase in the incidence of immune-related adverse events [3]. Metastatic CRC remains largely incurable, as CRC cells avoid immune surveillance and acquire resistance to immunotherapy through mutations in immune checkpoints and epithelial mesenchymal transition (EMT)-related genes [4]. Therefore, it is necessary to develop new therapeutic options for CRC.
Rapidly expanding expertise in immune surveillance and understanding of the underlying processes of immune evasion by tumors has resulted in the development of novel immunotherapy techniques [5]. The new treatments focus on overcoming immune system defects in response to tumor cells. Representative immunotherapies include adoptive cellular therapy, which involves the infusion of autologous immune cells such as dendritic cells (DCs), T cells, and natural killer (NK) cells. These approaches have yielded considerable improvements in preclinical and clinical trials for several cancers [6].
However, in solid cancers, immunotherapy has limitations, such as heterogeneity, insignificant infiltration, immunosuppressive tumor microenvironment (TME), and low cytotoxicity [7]. Since more than 90% of all cancers involve solid tumors, it is necessary to design and develop novel cellular immunotherapy strategies that can overcome the limitations of solid cancers. Several studies reported on the down-regulation of major histocompatibility complex class I (MHC-I) molecules on the surface of solid tumors [8]. We also confirmed the down-regulation of MHC-I in colon cancer in this study using imaging mass cytometry (IMC). Cytotoxic T cells can only recognize and kill cancer cells expressing MHC-I molecules that present their respective cognate antigens [9]. In the absence of MHC-I molecules on cancer cells, NK cells can recognize and kill cancer cells in an MHC-I antigen-presentation-independent manner [10]. This explains why single-cell-type therapy may be less effective than combined-cell therapy in eliminating cancer cells. In addition, the loss of MHC-I coupled with the immunosuppressive TME caused by the overexpression of immune checkpoint molecules and tumor heterogeneity is an immune escape mechanism [11].
The complementary interplay between immune cells can be an effective basis for cancer cell elimination. DCs, antigen presenting cells, interact with NK cells and these interactions are involved in NK cell activation, DC maturation, and immunoregulatory crosstalk [12]. NK cells are also engaged in the regulation of adaptive immune responses, especially the regulation of T-cell responses in various contexts [13]. Given this interplay, the need for combined immune cell therapy has emerged. A phase I clinical trial combining DC vaccination with adoptive T cell transfer in patients with stage IV melanoma was performed [14], and dual combinations of immune cells have been evaluated. However, triple combinations of the most potent immune cells (DCs, NK cells, and cytotoxic T lymphocytes [CTLs]) have not been evaluated.
Here, we investigated the combined tumor cytotoxic efficacy of primed DCs, CTLs, and NK cells against autologous cancer cells where our reported data suggested that the combination of DCs, NK cells, and CTLs eliminated cancer cells more effectively than single-cell therapies.
METHODS
Ethics statement
This study was approved by the Institutional Review Board of Seoul Songdo Hospital (No. 2018-007 and No. 2020-008). Samples were collected from 5 patients with CRC after obtaining written informed consent. Toxicity test was conducted in compliance with the Good Laboratory Practice Regulations by an external company (Dt&CRO Co Ltd) and was approved by the Non-Clinical Trial Center of Dt&CRO (No. 220045).
Sample collection
Patients with reasonable amounts of tumor and normal tissues obtained after surgery and who had provided consent for blood and/or tissue sample collection were included in the study. Blood (30–40 mL) was aseptically obtained from patients before surgery, and fresh tumor tissue samples were obtained immediately after surgery. Both tumor and normal tissue used in the study were confirmed by a pathology resident of Seoul Songdo Hospital (Seoul, Korea). The information of the patients participated in this study are shown in Table 1. The materials and reagents used in this study are outlined in Supplementary Table 1. All other reagents or equipment used in this study and not listed in the Supplementary Table 1 were of good manufacturing practice grade and are listed in the manuscript.
Imaging mass cytometry
IMC was performed using a Hyperion System (Fluidigm) as previously described [15]. Briefly, colorectal tissue slides from 111 patients were stained with hematoxylin-eosin, and a pathologist reviewed and marked the tumor/normal region in the slides, which represented the regions of interest for laser ablation. The slides were heated for 2 hours at 60 °C in a slide oven/dryer (Sejong Scientific) with a preheated antigen retrieval solution. The slides were then dewaxed in fresh xylene and washed with Maxpar water for 5 minutes. Slides were immersed in the preheated antigen retrieval solution and incubated for 45 minutes at 98 °C, leaving the lids loose. Slides were removed from the heating block and washed with water, followed by Maxpar phosphate-buffered saline (PBS) for 10 minutes with gentle agitation. The tissues were blocked with 10% bovine serum albumin (BSA) in PBS for 60 minutes at room temperature. The sections were incubated for 5 hours at 23 °C with an antibody cocktail in a hydration chamber (Simport Scientific) and washed with Maxpar PBS for 10 minutes with gentle agitation. Slides were then incubated with an antibody cocktail at 4 °C overnight, stained with Intercalator-Ir ((Fluidigm). The slides were air-dried for at least 20 minutes at room temperature (20–25 °C).
Twelve markers were detected through IMC, and multilayer images were obtained by transforming them to an OME-TIFF file using IMC data analysis software (MCD Viewer, Fluidigm). The antibody panels used are listed in Supplementary Table 1. In a single region of interest, among other cell types, tumor cells (pankeratin+), type 1 helper T (TH1) cells (CD3+CD4+CD8–Tbet+FoxP3–), cytotoxic T cells (CD3+CD8+), DCs (HLA-DR+CD11c+CD1a–), and myeloid-derived suppressor cells (MDSCs; HLA-DR–, CD11b+) were identified. Single-cell segmentation and the expression level of each marker based on OMD-TIFF files were analyzed using HALO (IndicaLab). After obtaining a positive cell counts, the cell count per 1 mm2 was evaluated, and the percentage of positive cells in the total cells was calculated.
Ex vivo NK cell expansion
Human peripheral blood mononuclear cells (PBMCs) were isolated from patient blood. Blood (10 mL) was slowly layered onto Lymphoprep (Axis-Shield Diagnostics). After density gravity centrifugation for 25 minutes at 1,224×g, a layer of mononuclear cells, or the buffy coat, was harvested. After washing the PBMCs with PBS, the PBMCs were co-cultured with 100 Gy γ-irradiated K562 feeder cells at a ratio of 1:6 (feeder cells to PBMCs) in Roswell Park Memorial Institute (RPMI) medium (Welgene) supplemented with 10% inactivated fetal BSA, penicillin-streptomycin, and 10 U/mL interleukin (IL)-2, 50 ng/mL IL-18, and 5 ng/mL IL-21. Every 2 to 3 days, half of the medium was replaced with fresh medium containing 20 U/mL IL-2 and 100 ng/mL IL-18. After 7 days, the cells were divided and supplied with fresh medium containing 200 U/mL IL-2 and 10 ng/mL IL-15 for 2 weeks.
Analysis of NK cell activating and inhibitory receptors
The expression of NK cell receptors was analyzed every 7 days using BD FACS Canto II (BD Biosciences). The cells were stained with fluorescence-conjugated anti-human monoclonal antibodies, including anti-NKG2D mAb-PE-Cy7, anti-NKp44 mAb-PE, anti-NKp46 mAb-PerCp-Cy5.5, anti-CD158b mAb-BV510, and anti-PD-1 mAb-PE. The cells were harvested, counted, and suspended in PBS containing 2% FBS (FACS buffer). Then, they were incubated with Fc receptor binding inhibitor polyclonal antibody at room temperature for 20 minutes. A total of 2×105 cells were seeded in 96-well U-bottom plates and stained immediately with the various antibodies at 4 °C for 30 minutes in the dark. After staining, the samples were washed twice with 200 µL of FACS buffer and transferred to a FACS tube. The Cytofix/Cytoper Fixation and Permeabilization kit was used for intracellular staining. The fixed/permeabilized cells were stained with antibodies (anti-CD152 mAb-PE Cy5) at 4 °C for 30 minutes in the dark. After staining, the cells were washed twice with FACS buffer. Analysis was performed using a BD FACS Canto II.
Tumor cell culture
The human colorectal cancer cell lines HT29 (ATCC HTB-38) and K562 were were purchased from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in RPMI medium supplemented with 10% FBS and 1% antibiotic-antimycotic (AA) in a 100 π culture dish in a humidified incubator at 37 °C with 5% CO2. For the primary tumor cell culture, tumor tissue was washed in sterile PBS supplemented with 0.5% gentamycin and 0.5% AA for 30 minutes, mechanically minced into small pieces (2 mm or smaller), and further digested using a tumor dissociation kit in a disposable gentleMAC C-Tube (Miltenyi), according to the manufacturer’s instructions. The digested samples were filtered through 70-µm strainers (Corning) centrifuged at 300×g for 5 minutes, and suspended in RPMI medium containing 10% FBS, 1×N2 supplement, 1×epidermal growth factor (EGF), 1% AA, and 1×Y27632.
Cytotoxicity of NK cells
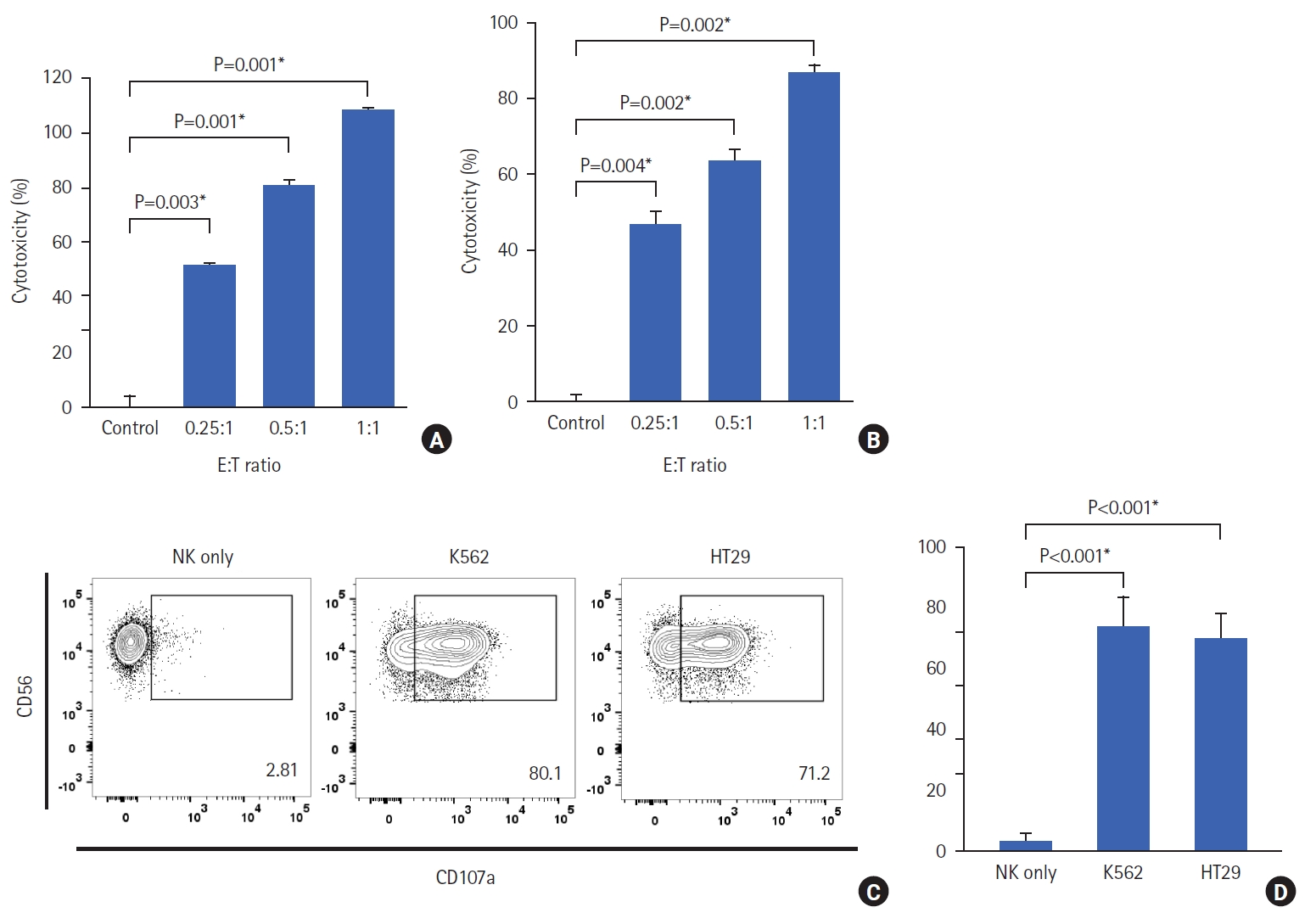
The cytotoxicity of NK cells was evaluated using a Cell Counting Kit-8 (CCK-8; Dojindo) assay, according to the manufacturer’s instructions. The target cells were K562 and HT29 cells. HT29 cells were prepared one day before co-culturing with effector cells (expanded NK cells). The effector cells were prepared in phenol red-free RPMI medium supplemented with 10% FBS and 1% penicillin-streptomycin antibiotics. K562 cells were co-cultured with effector cells at various effector to target (E:T) ratios (0.25:1, 0.5:1, and 1:1) at 37 °C in a 5% CO2 incubator. HT29 cells were also co-cultured with effector cells (0.5:1, 1:1, and 2:1) at 37 °C in a 5% CO2 incubator. Thereafter, 10 µL of CCK-8 was added to the mixed cells and incubated for 1 hour (K562) and 30 minutes (HT29). After incubation, the supernatant was harvested after centrifugation at 441×g for 3 minutes. Absorbance was measured at 450 nm using a FLUOstar Omega (BMG Labtech). NK cell function was evaluated using a CD107a degranulation assay. The K562 and HT29 cells were used as target cells. Effector cells were prepared with the same method as for the CCK assay. The effector cells were co-cultured with target cells (0.5:1) for 4 hours at 37 °C in a 5% CO2 incubator. After incubation, the cells were transferred to 96-well U-bottom plates and centrifuged at 2,000 rpm for 3 minutes. The supernatant was removed, and FACS buffer was added to the cells. The cells were stained with anti-CD3-FITC, anti-CD56-APC, and anti-CD107a-PE for 30 minutes at 4 °C in the dark. After staining, the cells were washed with FACS buffer and transferred to FACS tubes. Analysis was performed using a BD FACS Canto II.
Whole tumor lysate protein preparation
Whole tumor lysate protein extract was prepared by mincing fresh tumor tissue samples (0.1 g) obtained from the pathology department. The minced tissue samples were incubated in 1.5 mL Eppendorf tubes (Eppendorf) containing 2% HOCl in RPMI medium for 2 hours at 37 °C. The tissue samples were subsequently washed in 3 changes of cold PBS to remove residual HOCl. This was followed by protein extraction whereby 0.1 mL of EXB Extraction Buffer (Qiagen) supplemented with β-mercaptoethanol was added to the tube containing the tissue sample and mixed by vortexing. The tube was sealed with a clip and incubated on ice for 5 minutes. The tube was vortexed and incubated on a heating block (Lab Companion) at 100 °C for 20 minutes, transferred to a thermomixer (Eppendorf), and incubated at 80 °C for 2 hours with agitation at 750 rpm. Finally, the tube was cooled on ice and centrifuged for 15 minutes at 10,900×g. The supernatant containing the extracted proteins was transferred to a 1.5 mL Eppendorf tube. Detergent removal was performed using a HiPPR Detergent Removal resin-embedded Spin Column Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The tumor protein extract was stored at –80 °C until use.
DC preparation, whole tumor protein loading, and maturation
DCs were generated from CD14+ monocytes isolated from PBMCs using a 7-day protocol. PBMCs were separated from whole blood of the patients (30–40 mL) using Ficoll-Paque solution (Cytiva Life Sciences) and centrifuged at 800×g for 30 minutes continuously. At least 7×107 PBMCs were collected from the buffy coat layer and used for the isolation of CD14+ monocytes using CD14 microbeads and a Miltenyi autoMACS Pro Separator (Miltenyi), following the manufacturer’s instructions. The cells were cultured at a concentration of 1×106 cells/mL in DC medium supplemented with 1% streptomycin/penicillin, IL-4, and granulocyte-macrophage colony-stimulating factor (GM-CSF; 50 IU/mL each) in a 37 °C humidified incubator with a constant supply of 5% CO2. On day 3 of the culture, the medium was replaced with fresh medium containing IL-4 and GM-CSF (50 IU/mL each). On day 5 of culture, the medium was completely replaced with fresh medium containing 1% AA, and 50 IU/mL each of IL-4 and GM-CSF, to generate immature DCs (iDCs), or replaced with fresh medium containing 1% AA, and IL-4 and GM-CSF (50 IU/mL each) and 20 µg of whole tumor protein extract per 1×106 cells to generate mature DCs (mDCs). On day 6, the iDCs were matured by adding IL-1β (500 IU/mL), IL-6 (2,000 IU/mL), tumor necrosis factor (TNF)-α (860 IU/mL), interferon (IFN)-γ (20 ng/mL), prostaglandin E2 (PGE2, 40 μM), and CD40L (1 μg/mL) to the cells designated for mDC generation for 24 hours. On day 7, iDCs and mDCs were harvested for further analysis. Cell culture media were stored at –80 °C for further analysis.
DC phenotype analysis
iDCs or mDCs collected on day 7 of DC generation were suspended in PBS in FACS tubes. The cells were stained with a panel of phenotypic expression marker antibodies, including CCR7-PE, CD80-PE, CD86-BV421, CD83-APC, HLA-DR-PE-cy7, and HLA-ABC-PE. The cells were stained with the corresponding isotype controls and incubated at room temperature for 30 minutes, followed by washing with PBS prior to fluorescence detection. The stained cells were acquired using a BD FACS Canto II, and the cell population was recorded and analyzed using FlowJo ver. 10.7.2 (BD).
Migration assay
A DC migration assay was performed using a 24-transwell-plate. A total of 2×105 iDCs or mDCs were suspended in 400 μL of migration medium (DC media supplemented with 100 ng/mL GM-CSF and 100 ng/mL IL-4) and incubated in the upper chamber of the 24-transwell-plate. DC media (800 μL) containing 1 μg/mL CCL19 and CCL21 was added to the lower chambers. The plate was incubated for 16 hours at 37 °C and 5% CO2. Migration medium containing no CCL19 was added to the lower chamber as a control to detect spontaneous migration. After 16 hours of incubation, the cells from the lower chambers were harvested, and the cells were counted using a cell counting chamber and trypan blue. Cells that spontaneously migrated were subtracted from the migrated cells.
Measurement of cytokines
On day 7 of Mo-DC generation, cell supernatants were collected, and IL-12p70 was measured. After 4 days of co-culture of naive T cells and Mo-DCs, the supernatant was collected, IFN-γ was measured. Cytokines were measured using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems) following the manufacturer’s protocol without modifications. Optical density was measured using a Sunrise Microplate Reader (Tecan).
Isolation of naive T cells from PBMCs
Naive T cells were isolated from CD14– PBMCs obtained during the isolation of CD14+ monocytes for DC generation using the Naïve Pan T cell Isolation Kit (Miltenyi) and an autoMACS Pro Separator, following the manufacturer’s instructions with some modifications. In brief, CD14– PBMCs were suspended in 0.4 mL (per 107 cells) of autoMACS running buffer, and 0.02 mL (per 107 cells) of pan T cell biotin-antibody cocktail was added, mixed, and incubated for an additional 10 minutes on ice. Then, 0.03 mL of running buffer and 0.04 mL antibiotin microbeads were added per 107 cells, mixed, and incubated for an additional 20 minutes on ice. The negative fraction was collected as naive pan T cells. The cells were counted and seeded in T cell culture media containing 1% AA and 2000 IU/mL of IL-2 in 24- or 48-well culture plates until co-cultured with DCs.
DC and naive T cell co-culture and rapid expansion of activated T cells
On day 7 of DC generation, iDCs or mDCs were co-cultured with naive T cells at a ratio of 1:10 in 24- or 48-well culture plates (the negative controls consisted of naive T cells co-cultured with iDCs and T cells cultured alone). After 4 days of co-culture, microscopic images showing T cell proliferation were recorded using an Olympus IX71 inverted microscope (Olympus Global). The cell culture medium was removed and stored for cytokine analysis. Fresh antibiotic-supplemented T cell culture medium containing 6,000 IU/mL IL-2 was added to the wells to initiate pre–rapid expansion. Half of the medium was replaced with fresh antibiotic-supplemented T cell culture medium containing 6,000 IU/mL IL-2 at 3 day intervals. Depending on the proliferation rate of the T cells, the cells were transferred from the 48-well culture plate to 24- or 12-well plates or divided into 2 wells. After 14 days of pre–rapid expansion, the generated T cells were counted and used for cytotoxicity experiments.
DC, NK, T cell, and tumor cell co-culture (CCK-8 cytotoxicity assay)
Patient tumor cells were suspended in RPMI media at 1×105 cells/mL. The cells were seeded in 96-well flat-bottom culture plates (1×104 cells/100 μL) and incubated for 24 hours at 37 °C with 5% CO2. To preactivate the effector cells, 1×103 DCs, 5×105 T cells, 5×104 NK cells, and a 1×103 DC/5×105 T cells/5×104 NK cocktail were suspended in 100 μL of fresh RPMI media supplemented with 4,000 U/mL IL-2 and seeded in 1 well each of a 96-well U-bottom plate and labeled as DC/T, TC/T, NK/T, and DC/TC/NK/T corresponding to the E:T cells used. After 24 hours, the effector cells were suspended and centrifuged at 2,000 rpm for 3 minutes and the cell media was replaced with fresh RPMI media (100 μL). Next, the plate containing the tumor cells (target cells) was removed from the incubator, and the cell media of each well was replaced with 100 μL of cell media containing the effector cells. One well was left with only the target cells. The plate was returned to the incubator for 24 hours. Before the end of incubation, 10 μL of CCK-8 solution was added to the well containing only patient tumor cells to determine the color development time, which varied with different primary tumor cells. After determining the color development time, 10 μL of CCK-8 solution was added to the wells containing E:T cells and incubate at 37 ℃ in a 5% CO2 incubator for the predetermined time. When color development was complete, the culture plate was centrifuged at 2,000 rpm for 3 minutes, and supernatant were transferred to different 96-well flat-bottom wells to measure the absorbance at 450 nm using a FLUOstar Omega.
In vivo preclinical toxicity test
A toxicity test was performed using BALB/c nude mice intravenously administered with mDCs, CTLs, and NK cells twice at 2-week intervals. The low-dose group (G2) was administered 1×105 T cells, 1×104 NK cells, and 2×102 DCs. The medium-dose group (G3) received 1×106 T cells, 1×105 NK cells, and 2×103 DCs, and the high-dose group (G4) received 1×107 T cells, 1×106 NK cells, and 2×104 DCs. The control group (G1) was administered the same amount of PBS as the test groups. Repeated administrations were delivered twice at 2-week intervals.
The overall toxicity evaluation was accompanied by overall clinical pathology tests, including the observation of general symptoms, weight measurements, feed intake, and ophthalmological examination.
Statistical analysis
Significant differences between groups were determined using the Student t-test, and P-values of less than 0.05 were considered statistically significant. Data are presented as mean±standard error of the mean. For the statistical analysis of IMC, the percentages of marker expression were imported into R ver. 3.6.3 (R Foundation for Statistical Computing). Box plots were visualized with the ggplot2 ver. 3.3.2 (R Foundation for Statistical Computing).
RESULTS
Immunological phenotypes in colorectal tumor tissues
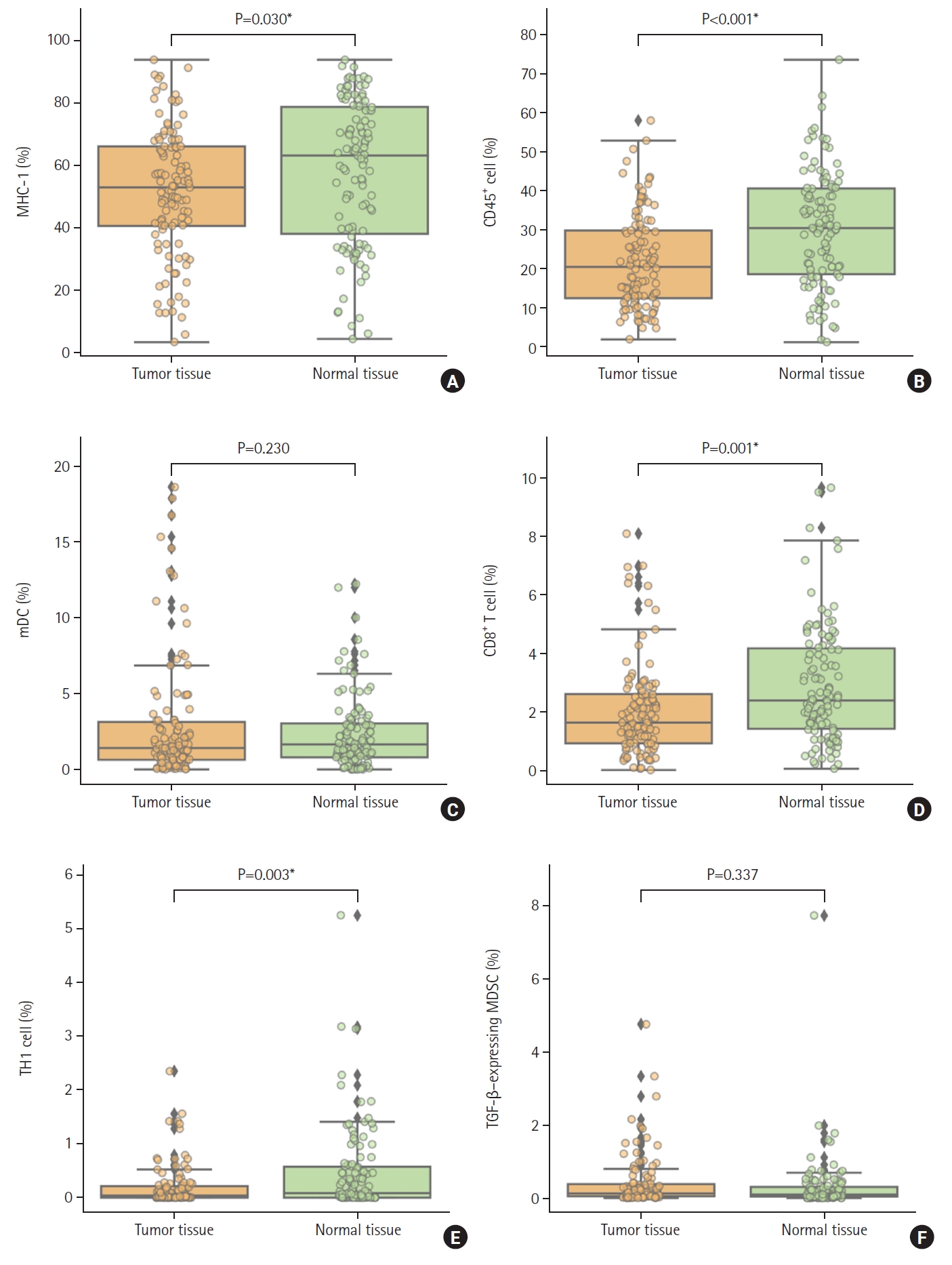
We investigated the immunological phenotypes in colorectal tumor tissues using IMC and compared them to those in adjacent normal peripheral tissues (n=111) as marked by a pathologist. MHC-I expression was significantly lower (P=0.030) in tumor tissues than in normal tissues (Fig. 1A). The expression of leukocytes (CD45+, P=0.001), CD8+ T cells (P=0.001), and TH1 cells (P=0.003) was significantly decreased in tumor tissues compared to normal tissues (Fig. 1B, D, E). In contrast, there was no significant difference in transforming growth factor-β–expressing MDSCs and mature DCs between tumor tissue and normal tissue (Fig. 1C, F).
Characterization of Mo-DCs
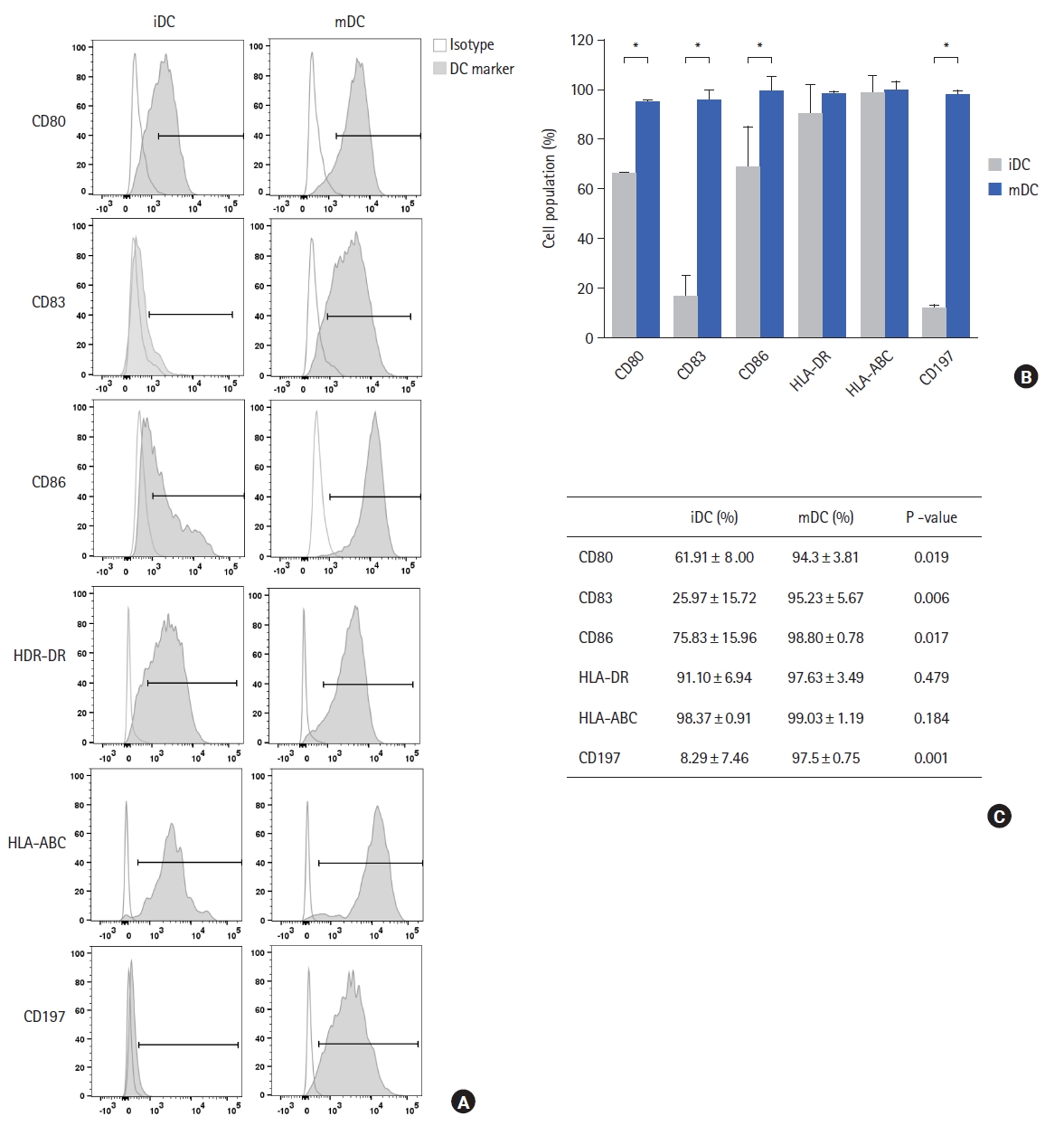
The expression of CD14 on iDC and mDC was significantly decreased on day 7 of monocyte differentiation demonstrating the complete differentiation of the monocytes. Compared to iDCs, mDCs expressed significantly high levels of CD83 (P=0.006) and CD197 (P=0.001) demonstrating that the mDCs were matured and capable of migration. Compared to iDCs, mDCs expressed significantly higher levels of CD80 (P=0.019), and CD86 (P=0.017), and CD40 which are co-stimulatory molecules, required for the activation of naive T cells. Other co-stimulatory surface molecules like HLA-DR and HLA-ABC, were equally expressed on iDCs and mDCs at higher levels, as expected (Fig. 2).
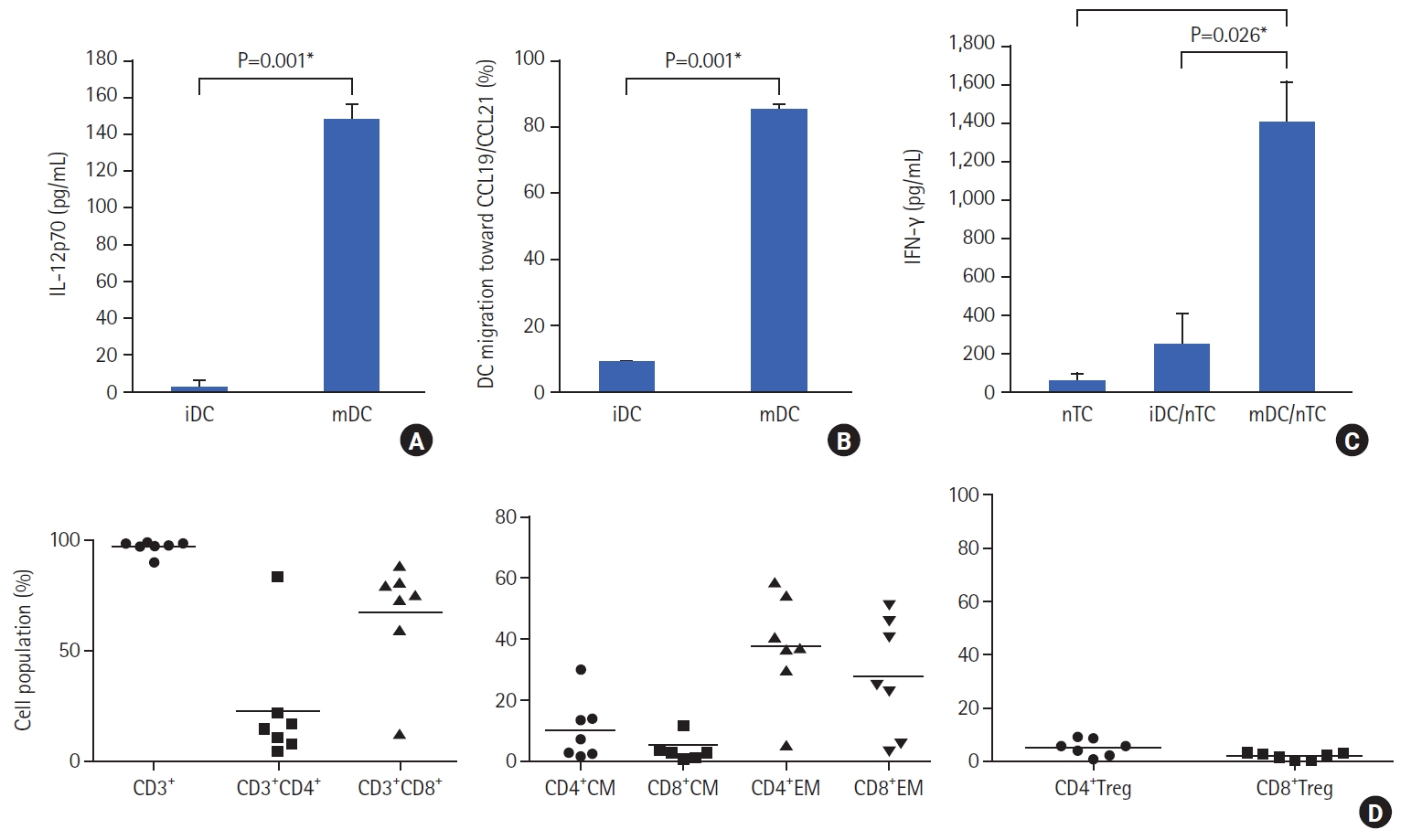
The secretion of IL-12p70, the bioactive form of IL-12, in the culture medium of iDCs and mDCs was analyzed. iDCs were generated from monocytes in the absence of tumor antigens or maturation supplements, whereas mDCs were generated from monocytes in the presence of tumor antigens and matured with a maturation cocktail that included IL-1β, IL-6, TNF-α, IFN-λ, PGE2, and CD40L. iDCs secreted very low levels of IL-12p70, whereas mDCs secreted significant levels of IL-12p70 (P=0.0004) (Fig. 3A).
For DCs to activate T cells, they must migrate from the periphery to lymph nodes to present antigenic peptides to T cells. The migratory ability test of iDCs and mDCs demonstrated that iDCs did not have the ability to migrate. In contrast, mDCs showed significantly higher (P=0.0004) migration ability toward CCL19/CCL21 chemokines (Fig. 3B).
Stimulation of T cells by Mo-DCs
The mDC stimulatory capacity of T cells was assessed and compared to that of iDCs using naive T cells. The activation of T cells was observed when mDC and naive T cells were co-cultured and this resulted in a significant increase (P=0.0086) in IFN-γ production (Fig. 3C). Further analysis of the activated T cells confirmed that 97.53%±3.12% expressed CD3, with 67.64%±25.81% of the CD3 T cells expressing CD8 and 23.16%±27.45% expressing CD4. Of the CD4 T cells, 10.03%±10.09% of the CD4 T cells were central memory T cells (CD197+ CD45RO+), while 5.23%±5.79% of the CD8 T cells were central memory T cells. Also, 37.51%±17.43% of the CD4 T cells were confirmed to be effector memory T cells (CD197– CD45RO+) and 27.49%±18.87% of the CD8 T cells were effector memory T cells. Of the CD4 T cells, 4.86%±3.03% were regulatory T cells (CD25+FOXP3+), and 1.74%±1.41% of the CD8 T cells were regulatory T cells (Fig. 3D).
Characteristics of cultured NK cells
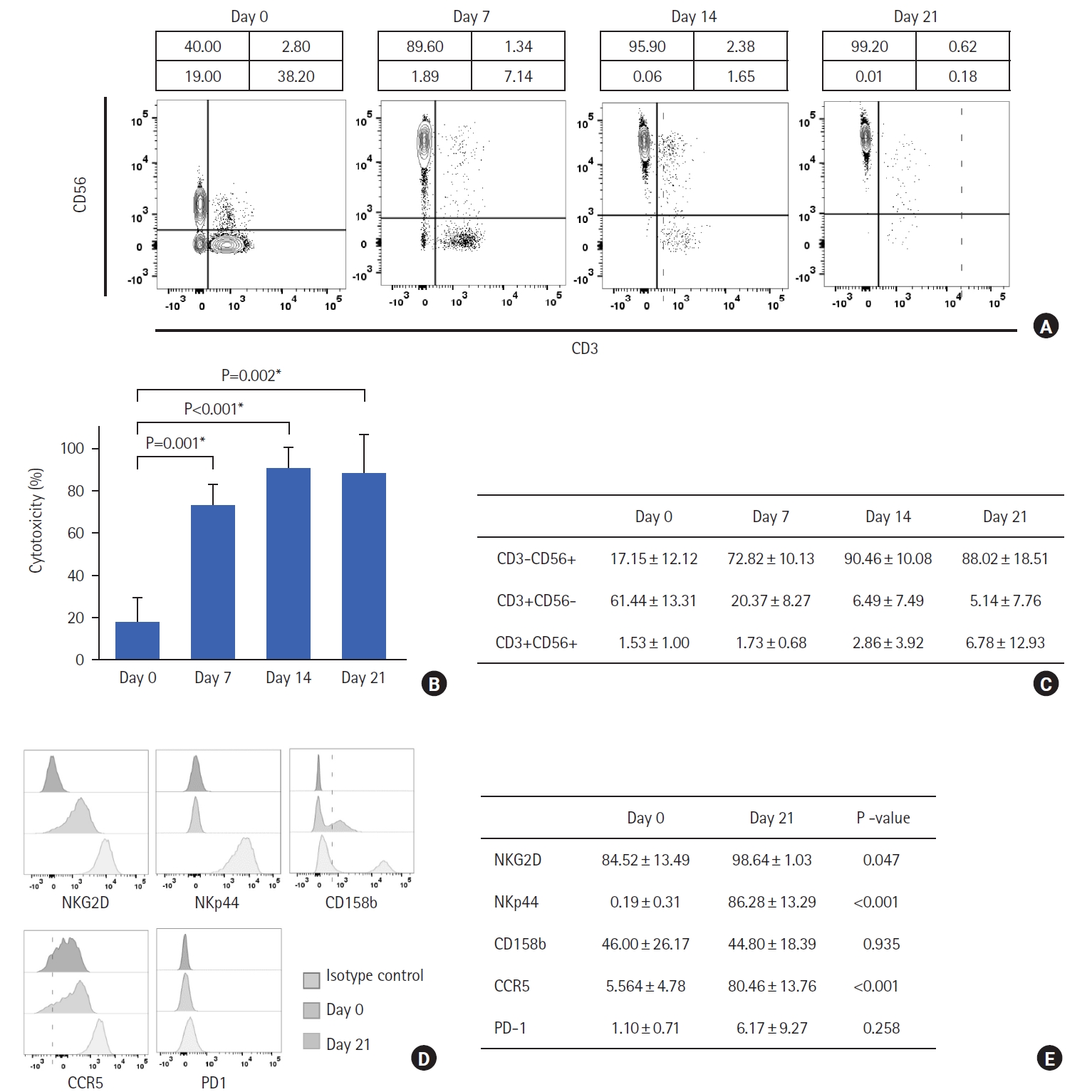
NK cells were expanded from PBMCs for 21 days. On day 0, the percentage of NK cells (CD3–CD56+) among the initially isolated PBMCs was 17.15%±12.12%. The percentage of the NK cells increased to 72.82%±10.13% on day 7 and to 90.46%±10.08% on day 14 and was slightly decreased to 88.02%±18.51% on day 21 due to variation in patient samples. The percentage of T cells (CD3+CD56–) in the initially isolated PBMCs was 61.44%±13.3%, which decreased to 5.14%±7.76% in the expanded NK cells by day 21 of expansion. The percentage of NKT cells (CD3+CD56+) in the initially isolated PBMCs was 1.53%±1.00%, which increased to 6.70%±12.93% in the expanded NK cells on day 21 (Fig. 4A). The expression of activating NK cell receptors in CD3–CD56+ NK cells (NKG2D, NKp44, Nkp46, and CCR5) and inhibitory NK cell receptors (CD158b) was analyzed in the initially isolated PBMCs on days 0 and 21. There was a significant increase (P=0.047) in the expression of NKG2D (from 84.52%±13.49% to 98.64%±1.03%) and NKp44 (from 0.19%±0.31% to 86.28%±13.29%) from day 0 to day 21. The expression of CD158b decreased from 46.00%±26.17% to 44.8%±18.39%. However, the differences were not significant. The expression of PD-1 increased from 1.11%±0.71% to 6.17%±9.27% but the differences were not significant. Also, there was a significant increase in the expression of CCR5 from 5.56%±4.78% to 80.46%±13.76% (Fig. 4B).
The cytotoxicity of expanded NK cells on the cancer cell lines (K562 and HT-29) was determined using the CCK-8 cytotoxicity assay. The CCK-8 cytotoxicity results showed that as the E:T cell ratio increased, cytotoxicity against K562 and HT-29 cells increased significantly (Fig. 5A). To understand the mechanism of action of NK cells in killing their targets, the cells were stained with CD56 and CD107a. There was no up-regulation of CD107a when NK cells were seeded alone. However, when NK cells were co-cultured with K562 or HT-29 cells, CD107a expression on the surface of the NK cells increased significantly (K562, 80.1%; HT29, 71.2%) (Fig. 5B).
Cytotoxic effect of combined immune cells against autologous cancer cells
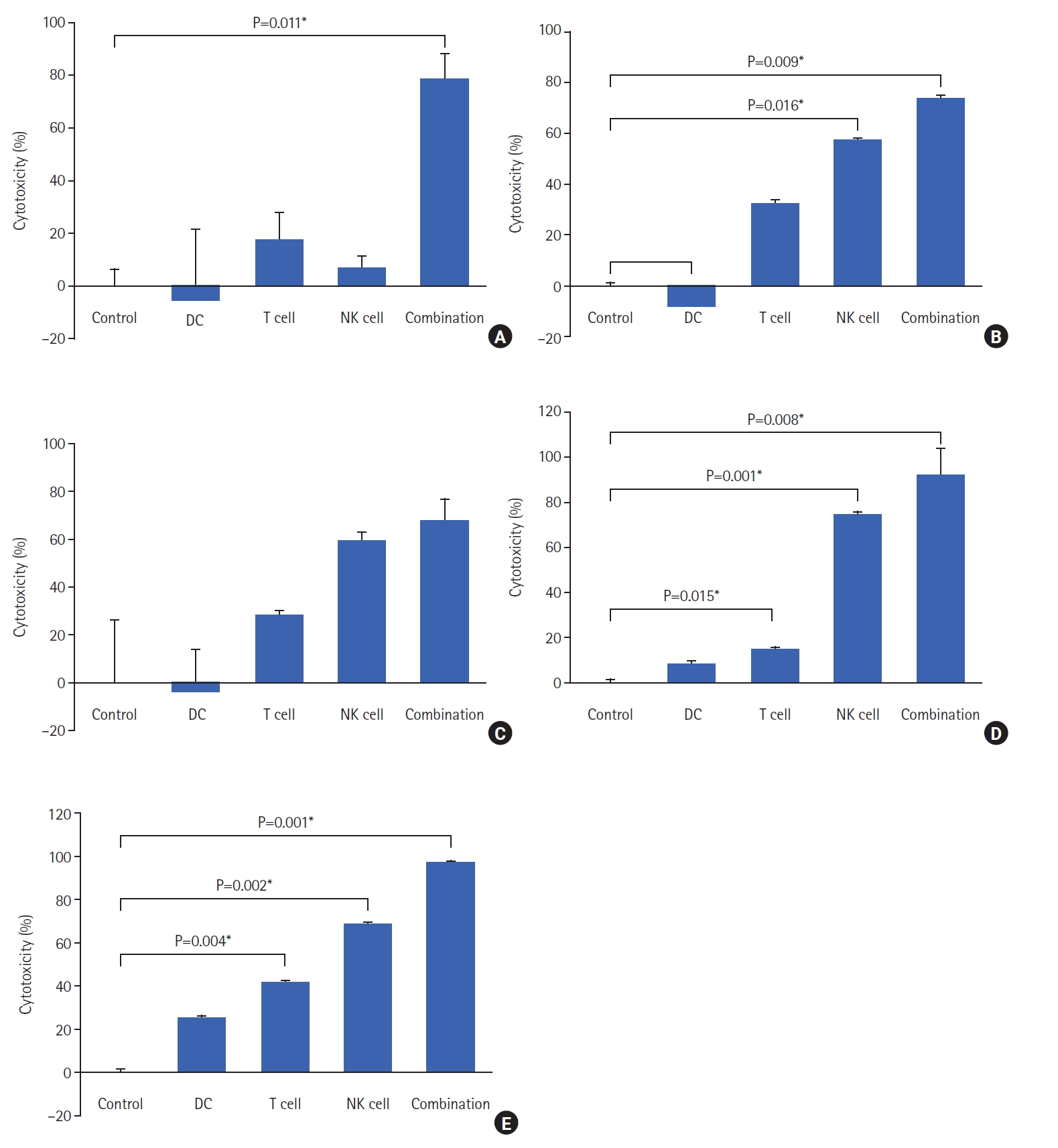
Our results were then validated by comparing the ability of Mo-DCs only, CRC Ag-specific CTLs, NK cells as single cells, and the combination of the 3 cells to kill autologous primary target cancer cells obtained from each donor. The combined immune cells with IL-2 were co-incubated with autologous cancer cells for 24 hours. After incubation, the viability of the autologous colorectal cancer cells was observed using the CCK-8 assay. In donor 1, when each single immune cell type was co-cultured with cancer cells, the cytotoxicity of T cells and that of NK cells were confirmed to be about 17.61% and 6.82%, respectively. However, the percentage of cytotoxicity from the combination of all 3 immune cells was significantly improved to 78% compared to controls. Combined mDCs, T cells, and NK cells significantly increased the cytotoxicity of autologous cancer cells in donor 4 sample (91.75%) and donor 5 sample (96.92%), compared to cancer cells cultured only with only mDCs, CD8+ T cells, or NK cells as single-cell treatments (Fig. 6).
Safety data to assess the possibility of clinical usage
To verify the toxic effect of the combined immune cell therapy reflecting for clinical usage, we performed single and repeated administration of the immune cells. One mouse in the low-dose group died on the 24th day of administration. An autopsy performed on the mice showed lysis in the brain and part of the abdominal organs due to spontaneous lesions and was not related to treatment administration (Table 2). Body weight, diet, and blood biochemical and histopathological tests were performed for each mouse, and no abnormal findings were identified according to the administration of the immune cell combination. Since nontoxicity was verified even with repeated administration of high doses of clinical doses, it could be concluded that safety equivalent to that of clinical dose was confirmed.
DISCUSSION
Clinical research for the use of ex vivo manufactured dendritic cells, T cells, or NK cells has been conducted for the treatment of various types of cancer. In the case of T cells, auto-cancer antigen-specific cytotoxic T cells that specifically recognize cancers cells have been used and exhibited antitumor effects. However, cancer antigens on cancer cells are not expressed in uniform rates and are continuously mutated. Also, the loss of MHC-I expression has been identified in some cancer cells. As a result, the antitumor effects of cytotoxic T cells alone can no longer be expected for cancers cells. This study was conducted to evaluate the ability of triple immune cell treatment involving autologous tumor lysate-loaded DCs, T cells activated by autologous tumor lysate-loaded DCs, and ex vivo-expanded NK cells for their ability to kill cancer cells. We proposed a combined triple immune cell therapy, which increased cytotoxicity toward autologous cancer cells, compared to single immune cell therapy (Fig. 6).
In the present study, a preliminary TME study involving 111 patients was performed (Fig. 1). It was confirmed that MHC-I was down-regulated in the TME as an evasion mechanism of tumor cells against T cells. In addition, the lower proportion of immune cells, such as CTLs, Th1 cells, and DCs at the center of the tumor compared to the invasive margin indicated the inability of immune cells to penetrate tumor tissues. This also implies that a certain number of immune cells are required to penetrate tumor tissues to eliminate cancer cells. These limitations may be overcome by using a suitable immunotherapeutic approach, including immune cell therapy.
DCs are a powerful tool for cancer immunotherapy, and their clinical efficacy is dependent on the type of antigen used to prime them. Whole tumor lysates are a promising source of antigens for DC vaccine therapy because of the presence of multiple immunogenic tumor antigens unique to individual tumors and natural adjuvants like heat shock proteins (HSPs) [16]. In this study, the whole tumor protein lysate generated by HOCL-oxidization was safe and non-toxic as administration of the tumor proteins did not affect the viability of the cells, thus demonstrating its feasibility in clinical application (data not shown). The treatment of tumor tissues with HOCl induces primary necrotic tumor cell death and presents immunogenicity-enhanced tumor protein antigens to DCs [17]. The DCs were successfully generated from CD14+ monocytes, and were capable of presenting antigens to T cells (Fig. 2). Upon maturation, DCs were shown to express high levels of CD83, the most specific marker for mature DCs, in addition to expressing co-stimulating receptors, such as CD80, CD86, HDL-DR, and HLA-ABC, which are necessary for the activation of memory and naive T cells. In addition, the mDCs demonstrated strong chemotactic migratory ability as the expression of CD197 was significantly up-regulated in the mDC and the cells showed great migration ability toward CCL19 and CCL21 chemokines. These results show that the mDCs produced were capable of migrating to lymph nodes to activate naive T cells and generate tumor-specific responses. Also, high IL-12p70 production by the mDCs confirmed the Th1-polarizing properties of the mDCs. Most importantly, it was observed that the mDCs stimulated naive T cells to rapidly proliferate and produce IFN-γ by CD8 and CD4 T cells. The activated T cells generated were predominantly CD8 T cells expressing lower numbers of regulatory CD8 T cells and central memory CD8 T cells but high numbers of effective memory CD8 T cells, thus demonstrating their ability to generate tumor-specific responses in vitro. The CD4 T cells contained lower numbers of CD4 T regulatory and central memory cells but higher numbers of effector memory cells.
NK cells play a crucial role in innate immunity and are valuable for cancer immunotherapy because they can recognize and kill malignant cells without prior priming or sensitization. We successfully expanded NK cells with high purity from PBMCs, that express high levels of activating NK receptors (Fig. 4). The cytotoxic ability of NK cells is strictly dependent on the balance between activating and inhibitory signals derived from receptors expressed on the cell surface [18]. The activating receptor NKG2D mediates target cell recognition and induces the production of IFN-γ, which plays a central role in tumor clearance [19]. NK cells expressing NKp44 recognize MHC class I and other related molecules and cellular ligands; this can induce NK cell responses [20]. Our NK cells were of high purity and showed an increased expression of activating receptors, such as NKG2D and NKp44, with no significant changes observed in the expression of CD158b inhibitory receptors. PD-1 was also expressed at low levels, indicating that the NK cells were not in their exhaustive state. The NK cells were also shown to express high levels of CCR5, indicating their ability to traffic to tumor regions. We performed an in vitro cytotoxicity assay using human colorectal (HT29) and K562 cell lines to further investigate the efficacy of the expanded NK cells. The NK cells exhibited a broad cytotoxic activity against both cell lines (Fig. 5).
Adoptive cell therapy based on DCs, T cells, or NK cells is highly effective for anticancer treatment [18, 21]. Immune responses are critical for the eradication of malignant tumors including colorectal cancer [21]. The presence of immune cells, such as NK cells, DCs, and cytotoxic T cells in the tumor microenvironment is extremely important for the eradication of tumors. This study evaluated the in vitro tumor cell killing ability of a cocktail of immune cells in the presence of IL-2 (DCs, T cells, and NK cells) that could reverse the immune evasion of autologous tumor cells (Fig. 6). The combination of DCs, T cells, and NK cells increased the cytotoxic ability of T cells and NK cells against autologous cancer, which was probably due to crosstalk among the immune cells. The results demonstrated that the combination of these 3 immune cells may be a more effective option for CRC immune cell therapy than the single immune cells therapy currently used in practice.
Developing an effective immunotherapy requires a complete understanding of the TME [22]. The TME contains a variety of immune-regulatory cells such as regulatory T cells (Tregs), tumor-promoting M2 macrophages, and granulocytic and monocytic MDSCs, all of which aid tumor progression by suppressing the in situ immune response [23, 24]. Adoptive cell therapy with a triple combination of NK cells, DCs, and DC-primed T cells could sufficiently substitute for the lack of immune cells at the center of the tumor. Due to the complexity of these combinations and the heterogeneity of immune cell subpopulations, additional in vivo experiments are necessary to evaluate and to predict their efficacy. Conducting in vivo preclinical studies using immunodeficient mice transplanted with human cancer cells or tissues, such as humanized mice, can validate the killing effect of the combination of the 3 cell types. Preliminary safety studies of the combine immune cells demonstrate no toxicity-related issues in mice.
In conclusion, these results suggest the possibility of combined-cell therapy using 3 immune cells fractions that significantly increased the killing of CRC cells in vitro. The present study was limited by its small sample size. Future studies involving larger sample sizes will greatly improve the significance of this study.