

INTRODUCTION
Ulcerative colitis (UC), a type of inflammatory bowel disease, is a chronic inflammation of the colon with an unknown etiology [1]. UC is an idiopathic disorder of the colonic mucosa which generally expanded proximally in a continuous manner through part of, or the entire, colon [2]. The clinical process is unpredictable, marked by alternating periods of exacerbation and remission [3]. UC is associated with morbidity, mortality, and substantial cost to healthcare; therefore, several studies have tried to identify the disease. The incidence and prevalence rates of UC vary genetic, environmental, and geographical factors [4]. UC is the highest incidence rates from industrialized countries, such as North America (6–15.6 per 105 people) and Europe (1.5–20.3 per 105 people) [4]. Patients with UC are relatively rare in Asia; however, they are gradually increasing in Asia [4] such as Korea [5], Japan [6], Hong Kong [7], and India [8] because of the westernization of lifestyles. In Korea, the mean annual incidence of UC was 1.51 per 105 people from 1986 to 2005. From 2006 to 2012, the mean annual incidence of UC increased significantly to 3.2 per 105 people [5].
UC is associated with an increased risk of colorectal cancer (CRC) [9]. Previously studies indicated a standardized CRC incidence ratio when compared with expected CRC incidence in the general population [10, 11]. UC-associated CRC only accounted for 0.15% to 5% of all CRC diagnosed in the general population; however, the risk of CRC has been reported to markedly increase with increasing probabilities of 2% by 10 years, 8% by 20 years, and 18% by 30 years [10]. Additionally, UC-associated CRC accounts for 10% to 15% of all UC patients’ mortality [9, 11]. In contrast to sporadic CRC, UC-associated CRC did not follow the typical “adenoma-carcinoma” sequences [2]. UC-associated CRC occurs as a result of repeated and prolonged inflammation in intestinal epithelial cells and has a sequence of “inflammation-dysplasia-carcinoma” from low-grade dysplasia and high-grade dysplasia to carcinoma [12, 13]. Dysplastic lesions occur in the form of 1 or 2 focal in the epithelial tissue where inflammation was induced, and pathological findings in the form of mucinous or signet ring cells are mainly observed [12, 13]. The main risk factors for UC-associated CRC include certain disease characteristics such as age at onset, extent and duration of disease, as well as non-UC characteristics such as family history of CRC [9]. However, excluding these factors, the biological factors and mechanisms of carcinogenesis have not been identified. Since limited understanding of the carcinogenesis of UC-associated CRC, the knowledge concerning the CRC risk in UC patients is still insufficient. Recent studies [14, 15] investigated UC-associated genomic alterations using targeted next-generation sequencing and reported certain discrepancy from UC-associated CRC in comparison with conventional CRC including higher frequency of TP53 mutations and lower frequencies of APC mutations.
In this study, we conducted a comprehensive analysis of 15 samples from 5 UC-associated CRC patients to understand the molecular characteristics of patients through a whole exome sequencing with tumor, dysplasia, and matched normal samples. To identify biological factors of UC-associated CRC, we analyzed mutation frequencies from tumor samples and biological function as well as generic alterations that were originated from earlier dysplasia status. Our study provides an in-depth understanding of UC-associated CRC and novel biological factors of carcinogenesis in UC-associated CRC.
METHODS
Patients and data collection
Five patients with surgically resected primary CRC were enrolled from Asan Medical Center (Seoul, Korea) between December 2014 and January 2017. The detailed clinical characteristics of the 5 patients are given in Table 1. Fifteen tissue sections were collected from recruited patients to identify somatic genetic variations in tumor and dysplasia samples, where tumor, dysplasia, and normal sections were collected from each patient’s formalin-fixed paraffin-embedded (FFPE) tissues.
This study was conducted with the approval of the Institutional Review Board of Asan Medical Center (No. 2018-0436) with a waiver for informed consent.
Whole exome sequencing of tumor, dysplasia, and matched normal samples
Genomic DNA was extracted from the FFPE samples and at least 1.0 μg of genomic DNA was used for constructing sequencing library. Sequencing library was constructed using the SureSelect Human Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA) by following the manufacturer’s instructions. Sequencing was performed on the NextSeq platform (Illumina, San Diego, CA, USA) with a read length of 150 bp, where tumor and dysplasia samples were sequenced with the average target depth of 200× while matched normal samples were sequenced with the average target depth of 100×. From the sequencing, 90.0% of sequenced bases showed quality scores higher than Q30.
Processing of sequencing data for calling somatic variants
Filtering of sequenced reads based on the base quality was performed using Sickle [16]. Filtered reads were mapped to the human reference genome (hg19) using Burrow-Wheeler Aligner ver. 0.7.12 [17]. The aligned reads were further processed for deduplication (using Picard ver. 1.119 [18]), local realignment and recalibrating base quality scores (using Genome Analysis Toolkit ver. 3.5 [19]). Somatic variants were called by Mutect2 ver. 3.5 [20] with default parameters. Somatic variations on exonic regions and splicing sites were selected for downstream analysis, while synonymous variations were eliminated. Driver mutation analysis was performed for dysplasia samples, dysplasia-initiated tumor samples, and The Cancer Genome Atlas (TCGA) CRC samples using DriverPower ver.1.02 [21], where the false discovery rate-adjusted q-score of < 0.1 was used as the threshold of driver mutations for the TCGA samples while low q-score of < 0.1 was used for the threshold of driver mutations for our dysplasia and tumor samples due to small sample sizes. In order to detect somatic copy number variations from dysplasia and tumor samples, EXCAVATOR2 [22] was used with default parameters.
Statistical comparison of somatic variations with generic colorectal cancer
The somatic variation frequencies of individual genes were computed from the tumor samples of 5 patients in this study, and they were compared with the mutation frequencies from the Catalogue of Somatic Mutations in Cancer (COSMIC) [23] database for CRC. If a gene showed difference in mutation frequencies with a P-value of < 0.01 by Fisher exact test, it was declared to have statistically significant difference in mutation frequency in dysplasia-initiated tumor samples in this study.
Functional annotation of genes with somatic variants
We selected the tumor mutation genes that have higher mutation frequencies from the 5 tumor samples than the frequencies from the COSMIC database [23] while showing statistical significance for the input of functional annotation. From the Molecular Signatures Database ver. 6.2 [24], 6,666 gene sets that represent canonical pathways, biological processes and molecular functions of Gene Ontology [25] were retrieved as reference of functional annotation. For each functional gene set, its overlap with the tumor mutation genes was computed, and the statistically significant P-value of overlap was evaluated based on the hypergeometric distribution. Functional gene sets that showed a hypergeometric Pvalue of < 0.01 were declared to have statistically significant associations with the tumor mutation genes.
We also evaluated the functional roles of the genes that showed somatic variations in both matched dysplasia and tumor samples. For the dysplasia and tumor samples from each patient, genes that showed the same somatic variations in both matched samples were declared as dysplasia-tumor common mutation genes for the patient. Each patient’s dysplasia-tumor common mutation genes were functionally annotated using the same procedure that was used for the annotation of the tumor mutation genes. Among the functional gene sets that showed statistically significant associations, we identified dysplasia-tumor common mutation-specific functional gene sets by using the following random permutation approach. In order to evaluate the possibility of functional association by random chance, we randomly built 10,000 lists of genes of the same amount based on the mutation frequencies from the colon adenocarcinoma data set of TCGA. Each of 10,000 random lists of genes was functionally annotated with the same procedure, and its associated functional gene sets were identified. If a functional gene set showed statistically significant association with the patient’s dysplasia-tumor common mutation genes and was also reported to have associations less than 100 of out 10,000 random trials (permutation P<0.01), the functional gene set was declared to be specific to the patient’s dysplasia-tumor common mutation genes.
RESULTS
Somatic mutational landscape of dysplasia-initiated colorectal tumors
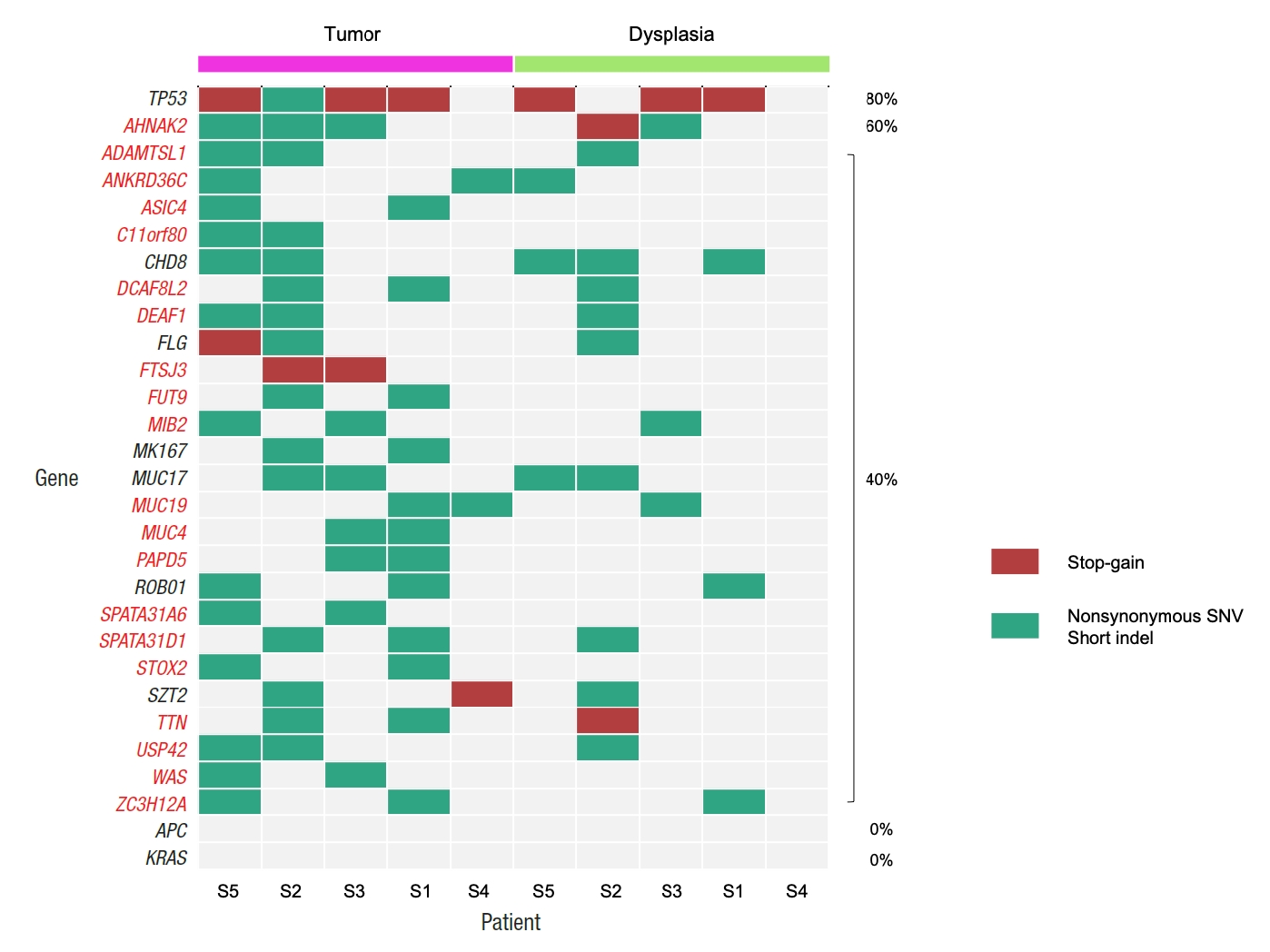
By comparing the mutation frequencies from tumor samples with the mutation frequencies that are reported in the COSMIC database, a total of 104 tumor mutation genes were identified with showing significantly different mutation frequencies (Table 2). All 104 genes showed higher mutation frequencies than the mutation frequencies from the COSMIC database. Fig. 1 illustrates the mutational patterns of selected genes among the 104 genes for matched tumor and dysplasia samples, where the mutation patterns of TP53, APC, and KRAS are shown together as they are frequently mutated genes in general CRC patients [26]. Table 3 also shows the mutation frequencies of TP53, APC, and KRAS. Four out of the 5 (80.0%) dysplasia-initiated tumor samples have mutations of the TP53 tumor suppressor gene, and this mutation frequency is higher than the mutation frequency of 31.0% that is reported in the COSMIC database even though the difference showed only marginal significance (P=0.0347). Three of the 4 TP53 mutations from the tumor samples were deleterious stopgain mutations, and the matched dysplasia samples with the 3 tumor samples also have identical TP53 stop-gain mutations. From the COSMIC database, APC mutations were reported with a frequency of 32.0% and KRAS mutations were reported with a frequency of 25.5%. However, none of the 10 tumor and dysplasia samples in this study showed mutation of APC or KRAS even though their frequency differences were not statistically significant. Regarding somatic copy number variations, no recurrent copy number variation was found from either dysplasia or tumor samples (data not shown).
Biological functions associated with dysplasia-initiated tumor mutations
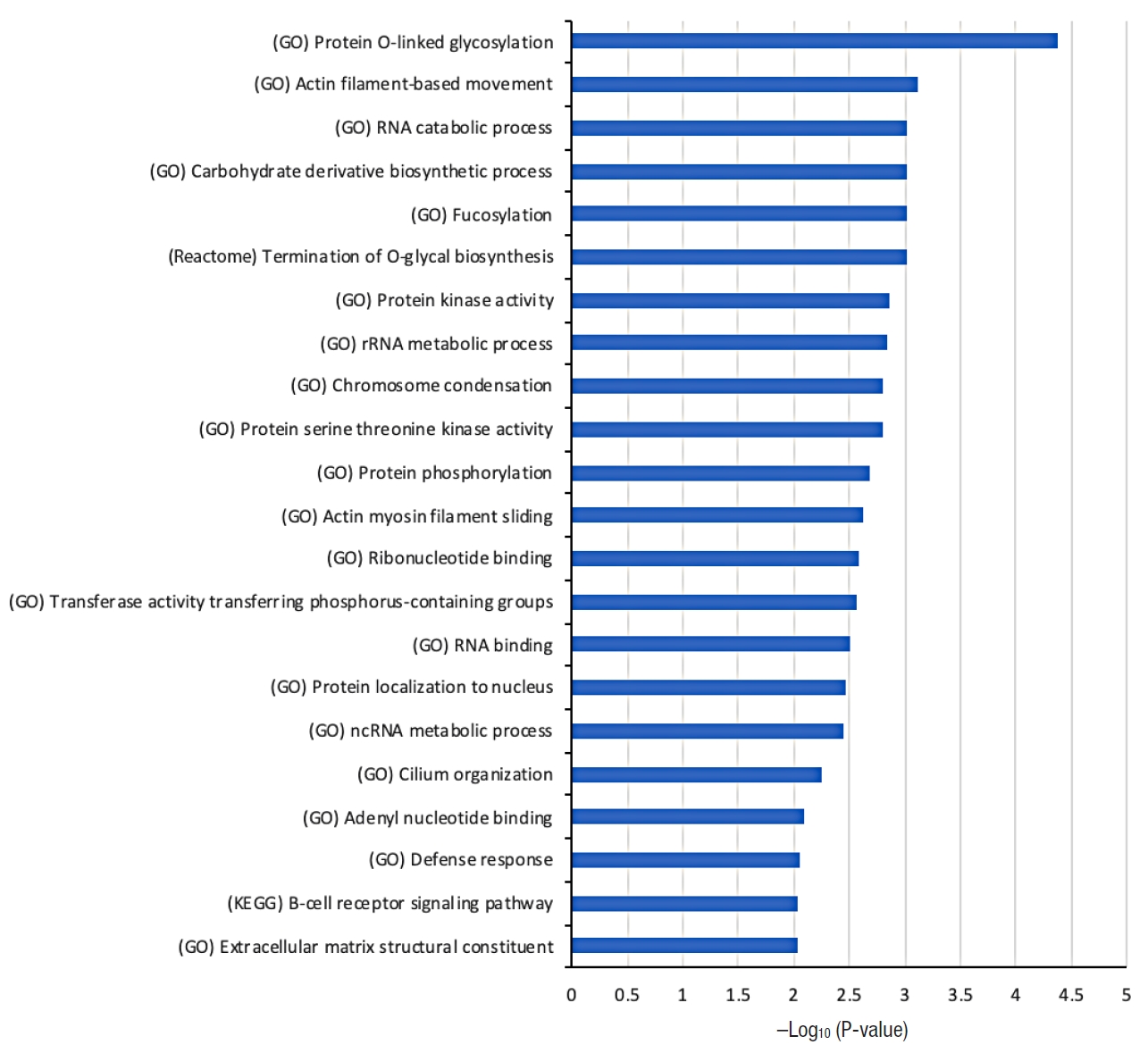
Fig. 2 shows the list of selected biological functions that are strongly associated with the 104 tumor mutation genes from the dysplasia-initiated tumors. Glycosylation-related functions (protein O-linked glycosylation and termination of O-glycan biosynthesis), functions related to maintaining cell structures (actin filament-based movement, actin myosin filament sliding, and extracellular matrix structural constituent), and immune-related functions (defense response and B-cell receptor signaling pathway) were strongly represented in the 104 dysplasia-initiated tumor mutation genes.
Somatic mutations of glycoproteins including mucins in dysplasia-initiated tumor mutations
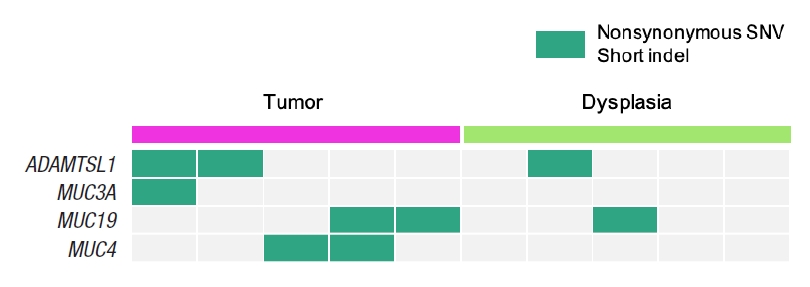
The most significantly associated biological function is protein O-linked glycosylation from Fig. 2, where 4 genes of ADAMTSL1, MUC3A, MUC4, and MUC19 from the 104 dysplasia-initiated tumor mutation genes are included. ADAMTSL1 encodes a secreted protein that resembles the members of a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS) family, while containing different domains including the thrombospondin type 1 motif. MUC3A, MUC4, and MUC19 are mucin genes that encode epithelial glycoproteins. Every dysplasia-initiated tumor sample has at least one mutation of these 4 genes (Fig. 3), and they are more frequently mutated in these dysplasia-initiated tumors than mutations in general CRCs from the COSMIC database (Table 4).
Common somatic mutations across matched dysplasia and tumor samples
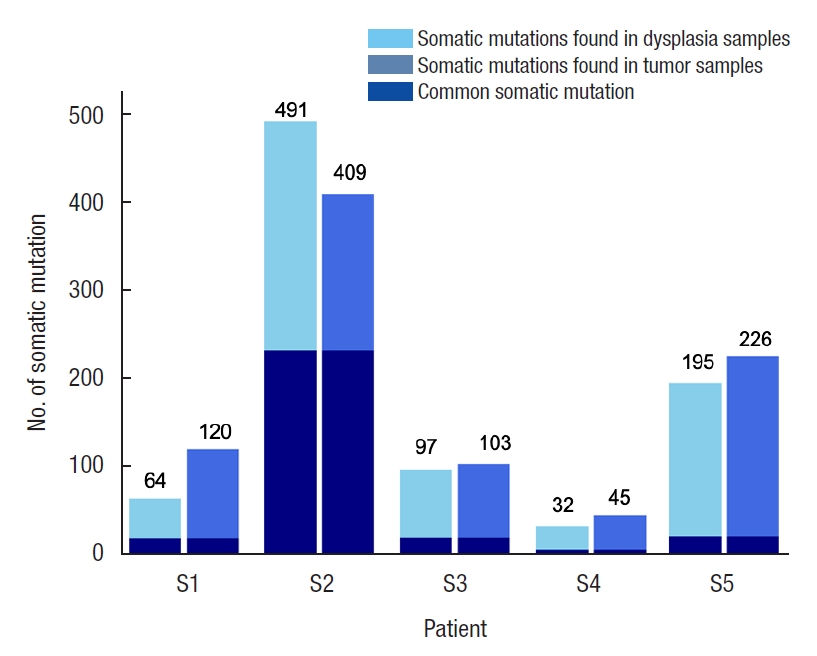
Table 5 and Fig. 4 show the number of somatic mutations from matched tumor and dysplasia samples for each patient, and the number of common mutations between the 2 tissue types. On average, 22.5% of somatic mutations in dysplasia-initiated tumor samples were already present from the matched dysplasia samples. These common somatic variations across dysplasia and tumor samples did not show clear patterns of increase or decrease in their variant allele fractions (Fig. 5). Among the common somatic mutations from the 5 patients, 3 patients (60.0%) have TP53 mutations and 2 patients (40.0%) have AHNAK2 mutations commonly in their matched tumor and dysplasia samples (Fig. 1). The dysplasia sample of S2 also has the same mutation with the matched tumor sample other than the illustrated stop-gain mutation shown in Fig. 1.
Dysplasia-tumor common mutation-specific biological functions
Table 6 shows the selected list of biological functions that are strongly associated with the dysplasia-tumor common mutation genes, where only the biological functions that are specific to these dysplasia-tumor common mutations are shown. Patients’ dysplasia-tumor common mutations show varying biological functions associated with them, while certain patients share similar functional profiles with their dysplasia-tumor common mutations. Biological functions related to stimulus response, apoptosis, NOTCH signaling, inflammation, and cell aging were associated with dysplasia-tumor common mutations from more than 2 patients.
Driver mutations analyzed from dysplasia and tumor samples
We investigated driver mutation genes from dysplasia samples and tumor samples. Four genes of C4BPB, SIGLEC7, TET3, and TP53 were identified as driver mutation genes from the dysplasia samples. From the tumor samples, 19 genes of AC016757.3, AHNAK2, ASIC4, ATP13A4, COL2A1, C11orf80, DCAF8L2, FLG, FUT9, HIF1AN, MUC17, NOTCH2, PLIN4, RAX2, SPATA31A6, SPATA31D1, SPNS3, TP53, and WSCD2 were identified as driver mutations. Only TP53 was a common driver mutation gene between the dysplasia and tumor samples. We also analyzed driver mutations for the 399 CRC samples with reported somatic mutations from TCGA, where 87 genes were identified as driver mutation genes; but they showed no common driver mutation gene with the results from our dysplasia and tumor samples (data not shown).
DISCUSSION
In this study, we investigated the somatic mutational characterization of 5 dysplasia-initiated CRC patients where mutational profiles of their tumors and matched dysplasia samples were analyzed. From the comparison with the mutational frequencies that are reported for general CRC in the COSMIC database, 104 genes were identified to show different mutation frequencies from the dysplasia-initiated tumor samples. All the 104 genes showed higher mutation frequencies compared to general CRCs, but a notable observation is the frequent mutation of the tumor suppressor gene TP53 (mutated from 4 of the 5 tumor samples) even though its mutation frequency was only marginally different from that of general CRC (80% vs. 31%, P=0.0347). More importantly, 3 of the 4 TP53 mutations were deleterious stop-gain mutations, and they were originally initiated from earlier dysplasia status (Fig. 1). This high mutation frequency and early establishment of somatic mutation from dysplasia status for TP53 strongly suggest that the aberration of TP53 plays an important role in driving dysplasia-initiated CRC. It is generally recognized that somatic mutations of other genes such as APC work as the main driver of initiating CRC, and the mutation of APC was reported with a frequency of 32% from the COSMIC database. However, none of the tumors or matched dysplasia samples in this study has APC mutation (Fig. 1). This is another evidence that dysplasia-initiated tumors have different tumor driving mechanisms with general CRC. Our result of driver mutation analysis also supports this point, where TP53 was the common driver mutation gene across matched dysplasia and tumor samples while no common driver mutation gene was identified from conventional CRC samples. Additionally, we could not observe any KRAS mutation from the tumor and matched dysplasia samples in this study (Fig. 1), while KRAS is often mutated in general CRC (25% from the COSMIC database) and causes resistance to several therapeutic agents. These findings of the important role of TP53 in UC-associated CRC and lower mutation frequencies of APC and KRAS are in concordance with previous studies [14, 15].
When investigating the biological functions associated with the highly mutated 104 genes from the dysplasia-initiated tumors, we observed multiple biological functions that can be related to dysplasia and inflammation, such as cell structure maintenance, and immune-related functions. We also observed strong aberration in glycosylation-related functions where it can be related to aberrated mucus formation on epithelial surfaces. Secreted extracellular proteins are often glycosylated, and it is known that epithelial tissue-produced mucins are heavily glycosylated. Mucins are major constituents of mucus, the viscous secretion that covers epithelial surfaces as those in colon. As glycoproteins including mucins play important roles in the protection of the epithelial cells, the aberration of glycosylation function suggests its strong correlation with the physiology of dysplasia-initiated CRC. We observed that 4 genes (ADAMTSL1, MUC3A, MUC4, and MUC19) are involved in this strong aberration in glycosylation, and we found that every dysplasia-initiated tumor in this study has mutations of at least one of these genes while general CRCs rarely have these mutations (Fig. 3). This suggests that dysplasia-initiated CRC can be associated with the disturbed ability to protect epithelial surfaces of colon due to mutated mucins and aberrated glycosylation function.
When we compared the somatic mutations of tumors and their matched dysplasia samples, we found that many somatic mutations already arose from dysplasia status, and a certain portion of them (22.5% on average) are carried to later established tumors. Biological functions that are specific to dysplasia-tumor common mutations include many functions related to cancer establishment and progression. The most frequently observed dysplasia-tumor common mutation was the mutations of TP53 (3 of 5 patients) and AHNAK (2 of 5 patients). TP53 is a well-known tumor suppressor gene, and it is noteworthy that its stop-gain mutations are established earlier from the dysplasia status as mentioned earlier in this section. AHNAK2 is an AHNAK nucleoprotein 2, and it may play a role in calcium signaling by associating with calcium channel proteins. The relevance between the AHNAK2 mutation and dysplasia-initiated CRC is not clear, but we will investigate its role in future studies.
We have analyzed the mutational characteristics and their importance in dysplasia-initiated CRC in this study by investigating somatic mutations of matched tumor and dysplasia samples from 5 patients. There have been previous studies that investigated genomic characteristics of UC-associated CRC using next-generation sequencing, and our result is in consistency with such studies. Nevertheless, our study can be distinguished with previous studies in 2-folds, where whole exome sequencing was used in our study to investigate entire genes while previous studies used only limited targeted-sequencing, and matched normal, dysplasia, and tumor samples were used in our study while only tumor tissues were analyzed in previous studies. One limitation of our study is the limited number of analyzed samples, as it is challenging to suggest stronger statistical evidence with 5 patients when comparing to general CRC populations. We aim to provide more sufficient analytical evidence in future studies with additional tumor and dysplasia samples.