INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder caused by germline mutation of the NF1 gene [1]. The NF1 gene encodes the tumor suppressor gene neurofibromin, and patients with NF1 are at an increased risk of developing various types of tumors, either benign or malignant [2]. However, there are few reports of NF1 associated with colorectal cancer. We present the case of NF1 in a 67-year-old patient with rectal cancer.
CASE REPORT
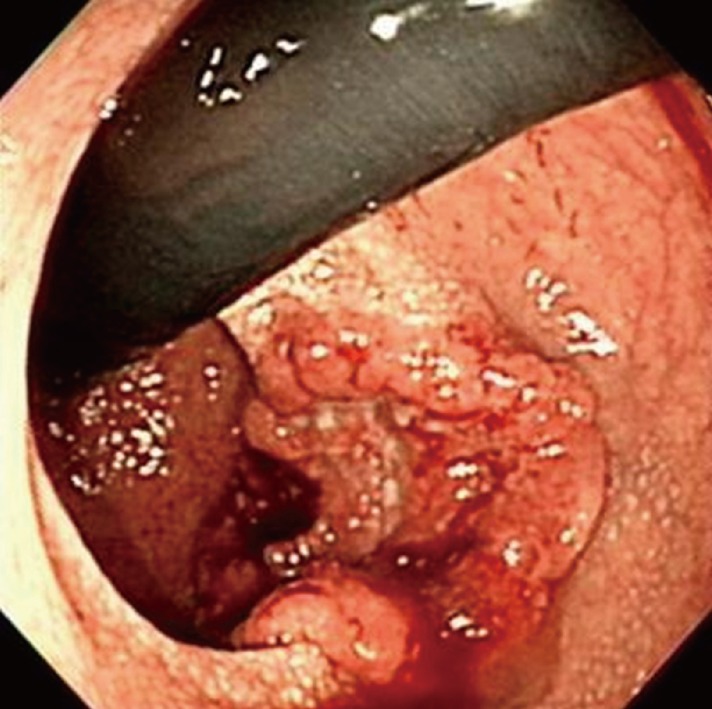
A 67-year-old man with a history of NF1 presented with hematochezia for 2 months. He had been diagnosed as having NF1 40 years earlier and had no family history of NF1. His skin showed numerous café au lait spots and cutaneous neurofibromas over the whole body (Fig. 1). An ulcerofungating mass was palpated at 5 cm above the anal verge on the posterior side of the rectum on manual rectal examination and was observed at colonoscopy (Fig. 2). The tumor was confirmed as an adenocarcinoma by colonoscopic biopsy. The abdomino-pelvic computed tomography (CT) scan revealed a 4.5-cm-long, irregular rectal wall thickening at the distal rectum, with a few small perirectal lymph nodes. A low anterior resection was performed, and the patient recovered uneventfully. The pathologic stage of the tumor was stage III (T3N1), and he received concurrent chemoradiation (6 cycles of 5-FU/leucovorin chemotherapy and 5,040 Rad of radiation).
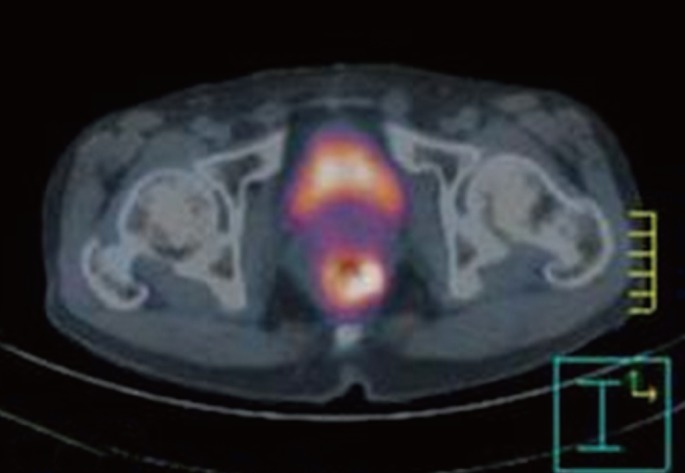
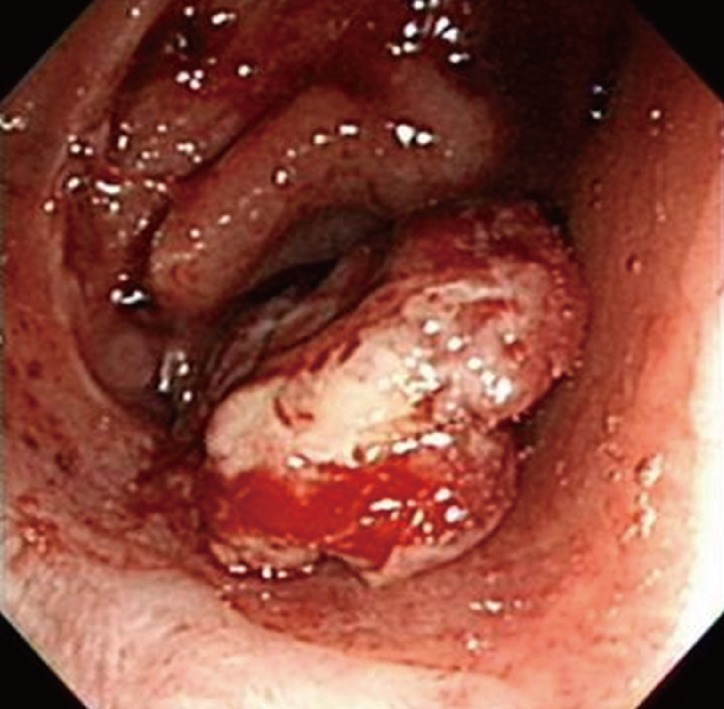
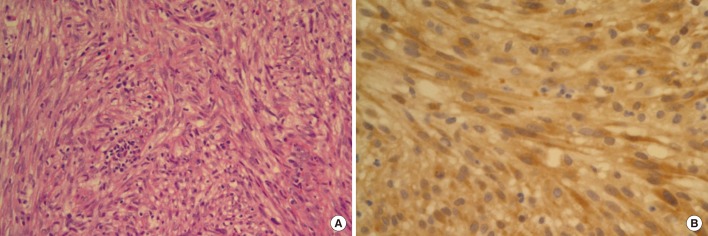
After completion of chemotherapy, he was followed up on 3 months interval. At 12 months after the operation, the positron emission tomography (PET)-CT scan showed increased fluorine-18 2-fluoro-2-deoxy-d-glucose uptake (SUVmax = 5.1) at the anastomotic site (Fig. 3). A 2-cm polypoid mass was found by colonoscopy and was excised transanally (Fig. 4). The tumor was composed of spindle cell fascicles of moderately atypical cells intermixed with a scanty amount of lymphoplasma cells (Fig. 5A). The tumor cells were positive to anaplastic lymphoma kinase and vimentin and were negative to CD34, CD68, S-100, SMA, and desmin (Fig. 5B). The tumor was diagnosed as an inflammatory myofibroblastic tumor (IMFT).
DISCUSSION
NF1 also known as von Recklinghausen disease, is an autosomal dominant disorder that affects one in 3,000 live births [1]. The syndrome results from inherited or spontaneous mutation of the NF1 gene. The most frequent clinical manifestations are skin pigmentation, iris Lisch nodules, and multiple cutaneous neurofibromas.
NF1 reduces the average life expectancy by 10 to 15 years, and malignant tumors are the most common cause of death in individuals suffering from this syndrome [2]. Malignant neoplasms in NF1 occur with an incidence of 3 to 15% and can arise in the nervous and the non-nervous systems in either childhood or adulthood. Malignant peripheral nerve sheath tumors and central nervous system tumors, such as the optic nerve glioma or astrocytomas are the most common tumors associated with NF1. Rhabdomyosarcomas and neuroblastomas are pediatric neoplasms common in children with NF1. Gastrointestinal stromal tumors, somatostatinomas, breast cancer, and pheochromocytomas are more common in adults with NF1 than in those without the syndrome. Development of epithelial malignancies is very rare, and the ontogenic relationship is not apparent. Adenocarcinomas in NF1 patients have been reported in the stomach, duodenum, small bowel, colon, and pancreas. In a 12-year follow up study in the Swedish NF1 population, 12 adenocarcinomas out of 70 patients (18%) were developed [3]. The authors stated that the incidence of adenocarcinomas seems to be high and that an adenocarcinoma is an important risk factor in NF1 patients. However, estimates of the frequencies of specific tumors in NF1 patients are difficult, and the incidence might be overestimated because most cohort studies are based on hospital data.
In the literature, there are only six case reports that indicate an association of NF1 with an adenocarcinoma of the colon [4, 5]. Nevertheless, considering that both NF1 and colon cancer are relatively common diseases, their concomitant occurrence is probably the result of a chance association. In a mortality study using US death certificates, colon cancer was not listed as often on the death certificates of persons with NF1 as on those of the general population [6].
Although malignancy constitutes an important cause of both morbidity and mortality in NF1, the mechanism underlying tumorigenesis remains obscure. Neurofibromin, the protein encoded by the NF1 gene, functions as a GTPase-activating protein for Ras by catalyzing the hydrolysis of active Ras-GTP to inactive Ras-GDP [2]. The NF1 gene may, thus, be considered as a tumor suppressor gene, and the high incidence of some types of neoplasms in NF1 patients may be related with mutation of the NF1 gene. It has been suggested that mutation of the NF1 gene can also occur in somatic cells and that loss of heterozygosity of the NF1 gene can be found in some sporadic colorectal cancers. One might expect that the tumor type found occasionally to harbor somatic mutations in the NF1 gene would have an increased incidence in NF1 patients. However, colon cancer does not seem to have an increased incidence among NF1 patients. There is a precedent for this paradox among hereditary neoplastic syndromes. Somatic mutations of the Rb gene are found frequently in sporadic small cell lung cancer, yet this tumor does not have an increased incidence among individuals with hereditary retinoblastoma. These considerations suggest that the NF1 mutation might promote growth of both the NF1-associated tumor type and other types of tumors by a somewhat different mechanism [7].
In addition to the uncommon concurrence of the two diseases, NF1 and cancer, there are some concerns about managing NF1 patients with cancer. Some NF1 patients have coexisting clinical problems that may complicate the clinical presentation, possibly delaying diagnosis and resulting in consequent presentation of the disease at a late stage. Multiple primary tumors, synchronous or metachronous, are more common in NF1 patients with cancer [8]. The initial cancer staging and the postoperative surveillance of NF1 patients may also be complicated by the presence of other benign or malignant lesions, visualized on imaging scans or identified on physical examination. In this case, an IMFT was mistaken as an anastomotic recurrence of the rectal cancer on both PET-CT and colonoscopy. Therefore, one should consider the possibility for synchronous or metachronous development of other types of tumors, besides tumor recurrence, when some tumors are found during follow up of NF1 patients with cancer.
In summary, we report an unusual case of rectal cancer in a patient with neurofibromatosis type 1. This case shows that a slightly different interpretation is needed for findings of initial staging and postoperative surveillance because multiple primary tumors are common in NF1 patients.